")
")
前回この原稿を書かせていただいたのは2021年であり、2年半ほどが経ってしまった。この間に、コロナが猛威をふるっていたが、この原稿を書くきっかけとなったmRNAワクチンのおかげもあってか何とか終息しつつあるように思える(実際はまだまだ広まっているのかもしれないが)。しかしそれよりも何よりも、その間に二人の偉大な先達を失ってしまった。キャップ構造発見者のお一人である古市泰宏先生と、私の指導教員でもある志村令郎先生である。怠けていたわけではないが、日々の教育・研究にかまけているうちに、お二人に読んでいただく機会がなくなってしまった。残念極まりない。もっとも、志村先生に読んでいただいた場合、「そんなことも考えてなかったのか!」とお叱りを受けそうである。編集幹事も甲斐田さんから小宮さんに代わられたが、小宮さんにも執筆をご快諾いただいた。もともと私の経験談(失敗談)を若い方達に聞いてもらうという趣旨だったので、気を取り直して書こうと思う。
(1)で書いたように、HeLa細胞核抽出液と32Pで標識した短いRNAを用いたゲル移動度シフト法を開発することができたため、まずはHeLa細胞のどの分画にキャップ構造への結合活性が多いのかを、HeLa細胞の核抽出液、細胞抽出液(S100画分)、リボソームウォッシュ画分を用いたゲル移動度シフト法で解析した。われわれが核内キャップ構造結合タンパク質を報告するまで、キャップ構造結合タンパク質は、細胞質に存在する、翻訳を司るeIF4Eタンパク質しか知られていなかった。HeLa細胞核抽出液をゲル濾過カラムで分画した実験から、HeLa細胞核抽出液中に存在するキャップ構造への結合活性は100 kD付近に存在することを見出していたため、分子量が約24 kDのeIF4Eとは異なると考えていたが、この実験により、私たちが見ているこのキャップ構造結合活性の80 %以上が核抽出液に存在することがわかり(図3)、eIF4Eとは別のキャップ構造結合タンパク質が存在することを確信した。

図3. キャップ構造結合活性の細胞内局在。図中Bと矢印で示すバンドがキャップ構造にタンパク質が特異的に結合しているバンドである。NE : 核抽出液、S100 : 細胞質抽出液、RW : リボソームウォッシュ画分。ほとんどのキャップ構造結合活性がNEに存在する。文献3より改変。
そして、核抽出液をMicrococcal Nuclease (MNase)で処理しても結合活性は失われないが、Proteinase Kで処理すると活性が消失したため、やはりこの活性はタンパク質因子の結合によるものであると考えた(図4A)。さらに、この結合活性の塩濃度への耐性を調べたところ、驚くべきことに1 MのKCl存在化でも結合活性が観察された。その塩濃度の条件では泳動が乱れてしまっているが、それでも結合は観察された(図4B)。別の実験であるが、キャップをつけたRNAからこの結合活性(タンパク質)を解離させるには、実に4 MのUreaが必要であり、強固な結合であることがわかった。このように、簡便な方法(例えば混ぜて流すだけ)と特異的で強い相互作用があれば、アッセイ系を確立しやすいということが言われるが、このケースは典型的な例だと思われる。

図4. 核内キャップ構造結合活性の生化学的検討。(A)では核抽出液をMNaseやProteainase Kで処理してからゲル移動度シフト法が行われた結果を示す。Proteinase K処理により、Bで示すバンドが消失する。(B)ではゲル移動度シフト法を行う際に、さまざまな異なる塩濃度を用いたものである。500 mM以上の条件では泳動が乱れてしまっているが、それでもBで示すバンドは観察されている。文献3より改変。
これらに加えてさまざまな条件検討の後、いよいよタンパク質の精製に取り掛かることになった。精製に使うHeLa細胞核抽出液の量は20 ml、総タンパク質量は468 mg/mlであった。ちなみに、核抽出液は浮遊培養しているHeLa細胞から調整するが、スピナーフラスコで6 Lを培養していた。そのうち1 Lを継代に用い、残りの5Lから核抽出液を調整する。一度に調整できる核抽出液は1.5〜2 mL程度であるため、まずは核抽出液を調整して-80 ℃で保存しておくところから始まる。その後、まずは陰イオン交換カラムであるDEAE-Sepharoseを用いて分画する。このカラムが直径2.5 cm、長さ18 cmという、通常よりかなり大きなカラムであった。こちらは自分たちで充填した。このカラムの0.15 Mと0.3 Mの間に溶出される画分を回収した。この後、ゲル濾過カラムにかけるのであるが、回収した画分の容量が大きくなっているため、75%飽和状態で硫安沈殿を行なって濃縮した。最近はあまり使われないかもしれないが、核酸のエタノール沈殿のように、タンパク質を変性させずに濃縮するのに非常に便利な方法である。その後、ゲル濾過カラムにかけるのであるが、このカラムも自分たちで充填を行った。カラムは直径が2.5 cmだが長さが92 cmもあり、白いゲル担体が充填された様子はさながら蛍光灯のようであった (図)。このカラムを詰める際にも、空気が入ってしまってやり直したり、下部の留めが外れて漏れたりというトラブルがあり、何度かやり直しを行った記憶がある。

それよりも先のDEAEカラムとこのゲル濾過カラムで大変だったのは、フラクションコレクターのトラブルである。フラクションコレクターは、分取する部分にセンサーが付いていて、何滴=何ml集めたら次に移り分取していく、という自動型のものであったが、分取するチューブの先がセンサー部分に触れやすく、滴数を正確に数えなくなることがあった。そこで、定期的に止めてチューブの先を見て、センサー部分を拭いてやらねばならない。10分間隔でやらなければいけなかったので、カラムとフラクションコレクターが入っている4℃のショーケースの前を離れられなかった。当時、スペースの問題でショーケースは廊下に置かれており、京都大学理学部生物物理学教室の廊下は、真冬は相当寒かった。タンパク質にはいいのかもしれないが、人間には寒すぎて、コートを着込んでいないと座っていられなかった。その様子を見かねた志村先生が、他のラボメンバーに、温かいコーヒーを持って行ってやれと言ってくださったのを覚えている。しかしそれでもやはり、大野さんも私も体調を崩してしまった。ここには書かなかったが、その前の条件検討のカラムがけも、4℃のコールドルームで行なっていた。コールドルームに入る時は、分厚い作業用コートを着るのだが、それでも寒かった。矛盾しているようだが、このコールドルームの作業が本当に辛いのは真夏である。外に出てきた時、30℃近い気温差で体がおかしくなる。本当に研究は体力勝負だなと思わされた。
話がそれたが、その後DEAEによる濃縮をし、キャップ構造の付いた短いRNA(ゲル移動度シフト法に用いるプローブと同じ)を固定化したSepharoseに結合させた後、0.7M KClによる洗浄を経て、4M Ureaで溶出を行い、最終的なタンパク質を得た。この辺りには、最初に行った条件検討が活きている。最近はこのようなハードコアの生化学を行うことはあまりないと思うが、なかなか経験し難い、有益な経験であったと思っている。最終的には、20 μg程度のタンパク質が得られた。SDS-PAGEでの泳動像も、80kD付近に単一のバンドがみられ、最終標品でもキャップ構造を持ったRNAへの特異的な結合が観察されたことから、精製がうまくいったと思われた(図5)。
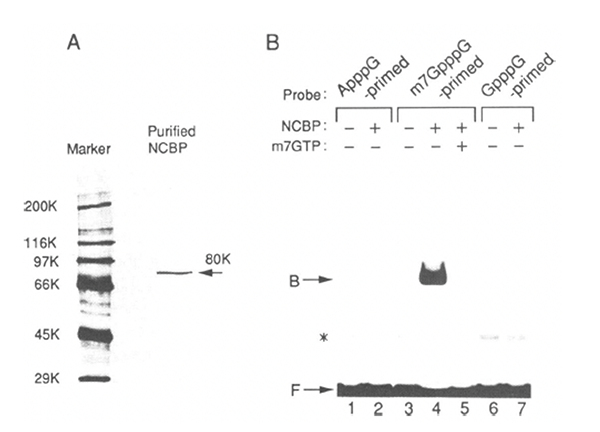
図5. 精製したNuclear Cap Binding Protein (NCBP)。(A)では70ng分を泳動した。分子量約80kDのあたりに単一のバンドが見える。(B)では、(A)に示した標品を用いてゲル移動度シフト法を示す。m7Gキャップ構造特異的なバンドが観察されている。文献3より改変。
大野さんが論文を投稿して受理され、あとはこのタンパク質のアミノ酸構造を決定する、という段階となった。現在ではヒトゲノム配列もすでにデータベースにあり、タンパク質はMS解析によってすぐに同定できるが、当時はタンパク質をトリプシン等の酵素で切断して出てきた断片を分取し、その断片の構造をN末側から決めていく、という手法をとっていた。そのため、大変腕の良い研究者にお願いしても、100 pmoleは必要、と言われていた。80kDのタンパク質なら8 μgである。今回のサンプルは十分量あったはずであったが、某大学に解析をお願いしたところ、全く何の情報も得られないままサンプルが無くなってしまった。そこでまた精製からやり直し、ということになった。これが私が大学院修士1年時の状況である。
(続く)
Ohno M, Kataoka N, Shimura Y. (1990)
A nuclear cap binding protein from HeLa cells.
Nucleic Acids Res. 18(23):6989-95. doi: 10.1093/nar/18.23.6989.