")
")
本稿では、ヒト培養細胞HEK293Tを用いた一過的高発現法による「リコンビナントタンパク質の調製法」を紹介させていただく。
I. はじめに
私は、mRNA監視機構に関わるタンパク質リン酸化酵素SMG-1の機能解析を15年にわたり続けている。この分子は3661アミノ酸の巨大酵素で、N末端側、C末端側両方に酵素活性必須領域が存在する。そのため活性を有する酵素を得るためには、全長タンパク質の発現が必要であった。ある程度予想はしていたが、大腸菌、バキュロウイルス、コムギ無細胞系などの方法でのリコンビナントタンパク質調製はうまくいかなかった。仕方なく、HEK293T細胞を用いた動物細胞タンパク質発現系の改良を続け、最終的に、化合物スクリーニングや、Cryo-EMによる立体構造解析が可能な量のリコンビナントSMG-1を精製可能な発現ベクター「pEFh」を樹立した(図1)[1, 2]。
このベクターと、近年廉価化が著しい人工遺伝子合成によるコドン最適化を用いることで、動物細胞培養を行う施設があれば新たな投資なしで簡便に全長のリコンビナントタンパク質を得ることが可能である。また、Plasmid transfection試薬として非常に安価なPolyethylenimine (PEI) ''Max'', (Mw 40,000) (Polyscience inc.)を使用することで、大幅にコストも低減されている[3]。
#もちろん、PUREシステム[4]、コムギ無細胞タンパク質合成システム[5]、ヒト細胞由来無細胞タンパク質合成システム[6]もおすすめです(すべて日本のRNA研究者産!)。興味のある方は調べてみてください。
II. ベクターの特徴
pEFhベクターは、N末端タグベクターでは1)共通のフレキシブルリンカー、2)HRV 3C proteaseによるタグの切り離し配列(HRV 3C site)、3)マルチクローニングサイトを有する。タグとして、SBP, Flag-SBP, HA-SBP, MBP, GST, sfGFP, Flag, HAを作成している。C末タグベクターは作成していない。タグを付与しないベクターも作成している(クローニングサイトはEcoRIのみ)。
これまでの高発現ベクターは、おもに転写量を最大化させることを主眼に開発されてきた(pSRα, pEF1, pCAG)。さらに、SV40 originやEBV oriPとそれぞれのエピソーマル複製因子であるSV40 large T antigenやEBNA-1を安定発現する細胞株(293T, 293FT, 293E)を用いることで、細胞内でPlasmidを増幅し、リコンビナントタンパク質の発現量を上げる(Large T: pSRα, pEF1α, pCAG, EBNA-1:pREP, pCEP)[7]。293E細胞は販売中止となっており、ベクター改良当時入手困難であったため筆者は検討していない。
pEFh vectorでは、mRNA転写後制御をターゲットとして、リコンビナントタンパク質の発現量増大を行った。TransfectionしたPlasmidからは、高発現のためのpromoterからだけでなく、いたる所から転写が生じている。また、高発現プロモーターから逆方向にも転写が生じている。これにより、二本鎖RNAが形成され、PKRの活性化依存的な翻訳抑制が起こる。さらに、SV40 polyA 配列による転写終結がうまくいかない場合は、RNA polymerase IIがPlasmidを一周し、新規の転写開始を阻害する。これらの問題を軽減するため、pGL4 plasmid(プロメガ)のベクターバックボーンを使用した。pGL4 ベクターバックボーンからは、ほ乳類の転写因子結合サイトが徹底的に除かれていることに加え(プロメガWeb site参照)、高発現プロモーター直前にα-globin転写終結配列が配置され、新規転写開始阻害を軽減する[8]。これにより、5倍程度のタンパク質発現量増加効果が得られた。アデノウイルス由来PKR阻害小分子RNAを発現するpAdVantage vector (プロメガ)をco-transfectionすることによっても、二本鎖RNA依存的な翻訳抑制は低減でき、タンパク質の発現量増大が期待できる[9]。さらなる二本鎖RNA発現低減配列を導入し、タンパク質発現量を増大させたpEFs vectorも作成している[10]。
eEF1α promoter、SRα promoter、CMV promoterの使用経験から、293T細胞では、eEF1α promoterが圧倒的に高発現であることを認識していた。そのため、eEF1α promoterを使用した。CAG promoter、Ub promoterについては検討していないが、293T細胞においてpEFh vectorが既存のpCAG vectorに比べ高発現であることは確認している。一部のCMV promoter vectorを含むこれらのプロモーターにはイントロンが含まれている。イントロンは、mRNA転写終結、核-細胞質輸送、翻訳といったすべての転写後制御過程を促進する。注意点として、発現するタンパク質のcDNA配列に3’スプライス部位配列がある場合、プロモーター中の5’スプライシング部位配列とのスプライシングにより、リコンビナントタンパク質発現低下が起こる可能性がある。これについては、cDNA人工合成により回避可能である。
上述の工夫に加え、mRNA分解抑制配列を3’UTRに(図2 lane1)、さらに、mRNA翻訳促進配列を5’UTRに(図2 lane 2)導入することで、それぞれ、5倍程度のリコンビナントタンパク質発現量増加効果が得られた。また、cDNAをコドン最適化し人工合成することで、2~10倍程度(遺伝子による)の発現増加が得られる(図2 lane3)。私自身は確認していないが、このpEFh vectorを複数の研究者に使用してもらったところ、特にRNA結合蛋白質についてはコドン最適化の効果が顕著であった。発現したRNA結合蛋白質が自身のmRNAに結合して発現量を抑えている可能性を考えているが、検証を行っていないので妄想の域を出ていない。最後に、N末タグの場合、cDNA由来の開始コドンはタグなしタンパク質を発現させる可能性があるため除いた方がよい。この発現ベクターで200種以上のタンパク質を試しているが、コドン最適化まで行った時点で、CBB染色による発現が確認できなかったことはない(精製できるかは別)。
抗体を用いないタンパク質精製タグとして、Strep tag II-、CBP-、SBP-、GST-、MBP-tagを検討した結果、SBPが最も、次いでMBPが回収率と精製度が高かった(それぞれについて、条件の最適化は行っていないので、あくまで参考程度)。CBP、GSTは動物細胞においてそれぞれの精製レジンに結合するタンパク質が発現しているため避けた方がよいかもしれない。His-tagについては、解析しているSMG-1やATMがNiやTALONレジンに結合してしまうため検討していないが、動物細胞からのタンパク質精製実績は豊富にある[7]。
III. プロトコール
■Plasmid construction
以前は、制限酵素、アルカリフォスファターゼ、TAKARAのDNA blunting kit・DNA ligation kit IIを駆使してPlasmid constructionを行っていた(いまでも、タグを替えるときや、Sequenceコストが無視できない場合に使用する)。今は、実験時間の節約と成功率UPが可能な、In-Fusion HD (TAKARA)やNEBuilder HiFi DNA Assembly Master Mix (NEB)を使用している。In-Fusion、HiFi DNA AssemblyどちらもChemical Competent Cellを使用した場合Transformationを強く阻害する。使用した結果、In-Fusionの方が影響が少なかったが、配列やPlasmidの大きさにもよると考えられる。3~6ピースのfragmentをligation後、直接Transformationすることもできるが、成功率は期待したほど高くはなかった。そのため、大きなタンパク質を人工合成する際には、コストの安い1000bp (GeneArt Strings: 18000円(Thermo Scientific))以下で合成し、DNAをHiFi DNA AssemblyでAssemble後、PCRを用いて増幅する(In-FusionではligationしたDNAにニックがあるため、PCRのテンプレートとして用いることができない)(図3A)。KOD FX Neoの登場でPCRがうまくいかないということはほとんどなくなった。しかし、結構な頻度(3000bpに1塩基くらい)で変異が入るため、作成したPlasmidのSequenceは必須となる(あくまで個人的感想)。
材料
- 制限酵素(SmaI, EcorV, PvuII, DpnI)
- KOD FX Neo DNA polymerase(TOYOBO), 3000bp以上のPCR
- PrimeSTAR Max DNA Polymerase (TAKARA), 2000bp以上3000bp以下のPCR
- iProof DNA polymerase (BioRad), 2000bp以下のPCR
- In-Fusion HD cloning kit (TAKARA)
- NEBuilder HiFi DNA Assembly Master Mix (NEB)
- AMPureXP (ベックマンコールター)(DNA溶液のバッファー交換、濃縮に使用)
- Wizard SV Gel and PCR Clean-Up System (Promega)、FastGene Gel/PCR Extraction Kit (安い! 日本ジェネティックス)など
- コンピテント大腸菌、DH5α、NEB stable(上手くいかない場合)など
- DNAミニプレキット, NucleoSpin Plasmid EasyPure (TAKARA)、FastGene Plasmid Mini (安い! 日本ジェネティックス)など
- DNAミディプレキット,NucleoBond Xtra Midi (TAKARA)など(293T細胞はTLR4の発現が低いため、エンドトキシンFreeの必要性は低い)
- NucleoBond Xtra BAC (TAKARA)(精製が困難なPlasmid、15kbp以上の大きなPlasmid)
*制限酵素、PCR酵素などは、ベンチトップクーラー(おすすめ器具参照)に入れた状態でフリーザーに保存することで、長期間保存による失活を防ぐ。
*チップ、チューブはオートクレーブなどで滅菌する必要なし。
おすすめ器具
- アイスオン(エスケーバイオ)
- NEBクーラー(NEB)、Biocooler (BioSmith)、DyNA Chill(Labnet)などのベンチトップクーラー
方法
1. マルチクローニングサイトを制限酵素、もしくはインバースPCRにより開裂させることによりベクターを作成する。インサートをPCRで増幅しない場合、インバースPCRのプライマーにはインサートの5’、3’それぞれに15bpの相同配列を持たせる。
2. インサートDNAをPCRにより増幅する。プライマーは開裂させたベクターの5’、3’それぞれに15bpの相同配列を持たせる(図3B)。HiFi DNA Assemblyによるインサートのアセンブルを行う場合、各0.5 μL (10-25 ng)のfragmentを1.5 mLチューブに加え、等量の2x HiFi DNA Assembly master mixを加える(マニュアルの1/10スケール~、160円~)。50oC、15分反応後、AMPureXPにより精製、~30 μLの超純水で溶出。溶出後、約半分の溶出液をテンプレートにアセンブルした全長をPCRにより増幅。
3. Ampicillin耐性plasmidがテンプレートの場合、PCR後、DpnIによりテンプレートDNAを消化する。DpnIは大腸菌でメチル化されたDNAを選択的に切断する。制限酵素で開裂させたplasmidはDpnIで消化されるので注意。
4. ベクター、インサートをそれぞれアガロースゲル電気泳動により分離、精製する。
5. 各0.5 μL (10-25 ng)のベクター、インサート(合計1 μL)と0.25 μLの5 x InFusion HD(コントロールは滅菌水0.25 μL)をシリコナイズチップで1.5 mLシリコナイズチューブ(サーマルサイクラーを使用する場合は0.2 mL PCRチューブ)に加え、ピペッティングにより攪拌する(マニュアルの1/8スケール~、300円~)。50oC、15分反応後、氷上に移す。
6. 25 μL以上のコンピテント大腸菌を直接ligation反応後のチューブに加え、そのままピペッティングにより攪拌し、1~5分氷上で静置。
7. 42oC、1分間保温後、氷上に移す。0~75 μLの滅菌水を加え、アガロースプレートに塗る。アガロースプレート節約のため、マジックで半分~5つの領域に分けて塗る。4つ以上に分けた場合、滅菌水は加えない。
8. 一晩培養後、コントロールligationのコロニー数を上回っている場合、2~3コロニーを培養。(急ぐ場合はKOD FX Neo (0.1 μL/tube)とシークエンスプライマーを用いてコロニーPCR (total 15 μL)によりベクターと連結したインサートを増幅し、そのままSequence。)
9. ミニプレキットによりplasmidを精製し、制限酵素により構造を確認。Sequence(インサートにPCRを用いた場合必須)。急いでいる場合、293T細胞を用いて発現確認用のPlasmid transfection。
10. Sequence確認後、DNAの大量精製を行う。
■Plasmid transfection
材料
- 293T細胞(ATCC) (もしくは293FT細胞:Thermo scientific)
-
pEFh vector (試したい方はリクエストしてください。
This email address is being protected from spambots. You need JavaScript enabled to view it. ) - PEI: Polyethylenimine ''Max'', (Mw 40,000) (Polyscience inc.) (1μg/mLとなるようDistilled waterで溶解。NaOHでpH7.0に合わせる。25円/mL。1~1.2mL/1.5mL tubeに分注し、-20oCで長期保存。凍結融解でTransfection効率が約半分に低下するため、一度溶かしたら、4oCで保存。4oC、3ヶ月の保存でTransfection効率はほとんど変わらない。私自身はまだ試していないが、siRNA transfectionには分子量の異なるPEIが適しているらしい[11]。)
- 10%FCS/DMEM培地
- PBS(-) (室温)(Opti-MEM(室温)にしてもよい)
- (PLL: Poly-L-lysine solution, 0.01% (P4707, Sigma))
方法(15 cm dish 1枚分)
1. Option(293FTでは推奨): 実験1日目、朝、15cm dishに5mlの0.0005 % [1:20] PLL (滅菌水で希釈、用事調製) を加え1時間室温静置。静置後、溶液を完全に取り除き数時間乾燥させる。
2. 実験1日目、昼、培地を37oCにあたためる。PBS(-)は前日から室温。
3. Plasmid 15ugを1mL PBS(-)(室温)に加え、vortexにより攪拌。
4. 75 μL PEIをPlasmid/PBS(-)溶液に加え、vortexにより攪拌。攪拌後室温で30分静置。
5. 1.3x 107 cells/15 cm dish(PLLコート)/18mL 培地となるよう細胞を播種。
6. 手順4のPlasmid/PEI/PBS(-)溶液を手順5の細胞に加える。細胞の接着を待つ必要はない。
7. Option: 培地節約の必要がない場合や強い細胞毒性が観察された場合、Plasmid Transfection後6~8時間後に培地交換。培地交換を行う場合、PLLコートdish使用を推奨。
8. 実験2日目の夕方~3日目夕方(細胞回収の16時間前~)、5 mL 培地を加える。293FTの場合、培地を加えるだけで細胞がはがれる場合がある。(栄養飢餓による翻訳効率低下を避けるため。eEF1α promoterは栄養飢餓依存的翻訳低下を受ける5'TOP (Terminal Oligo Pyrimidine motif) 配列を有するmRNAを転写する[12])
9. 実験2日目夕方~4日目朝(Plasmid Transfection後30~70時間後)細胞を回収。そのまま精製に用いるか、ペレットの状態で凍結保存。通常は44~48時間後。毒性の高いタンパク質や分解されやすいタンパク質の場合30時間前後。安定なタンパク質であれば、70時間前後。GFP, RFP, mKate2などの可視光に励起波長をもつ蛍光タンパク質であれば、蛍光灯下でCellペレットの蛍光が観察できる。
■SBPタグを用いたタンパク質精製
さまざまなメーカーのStreptavidin sepharose/beadsを検討した結果、タンパク質の大量精製にはGE health scienceのStreptavidin sepharose high performanceが最も優れていた(個人的には)。10年ほど使っているが、Lotによりバックグラウンド吸着の強いものがあるので、レジンブロッキングは必須(個人的な感想)。Mag sepharoseを使用すると精製度は上がり、実験の手間を省くことができるが、高コスト。全長タンパク質であれば多くのRNA関連分子の場合可溶性に問題はない。一方で、タンパク質の部分配列を高発現した場合、可溶化しないことがある。短い領域の場合は、タグをMBPにするとよい[2]。MBPの二量体形成が気になる場合は、SBP-sfGFPをタグとする。紹介する条件で可溶化しない核酸結合蛋白質の場合、400mM NaClにすることで可溶化できたことがある。ミトコンドリアに局在するaminoacyl-tRNA synthetaseの可溶化はうまくいかずギブアップした。細胞に高発現しすぎることで凝集体を作るような場合も工夫が必要となる。また、可溶性に問題がなくても、レジンから溶出できない場合がある。溶出バッファーの工夫で溶出できたとしても、保存中に活性が失われる場合が多い。このような場合、On beadsで酵素反応や結合実験を行う。On beadsでの実験を行う場合SepharoseではなくMag beadsを使用することを勧める。私が使用しているのは、Dynabeads M-280 Streptavidin (for SBP)、Anti-HA-tag mAb-Magnetic beads (TANA2, MBL)、Anti-DDDDK-tag mAb-Magnetic beads (FLA-1, MBL)。SBPタグで精製したタンパク質同士の結合実験のために、Baitとなる分子にFlag-SBPかHA-SBPタグを用いる[10, 13]。
材料
- SBP融合タンパク質を発現したCellペレット
- Streptavidin sepharose high performance (GE health science)
- Streptavidin Mag sepharose high performance (GE health science)(より高い精製度が必要な場合。溶出volumeを減らしたい場合。)
- d-Desthio-biotin (D1411, Sigma)(節約する場合、透析が不要な場合はBiotin)
- PI: Protease inhibitor cocktail (ナカライ)
- PPI: Phasphatase inhibitor cocktail (ナカライ)
- 防腐剤: ProClin 300 (Sigma-Aldrich)
- QIAshredder (QIAGEN)(タンパク質可溶化のため、lysis bufferの塩濃度を上げた場合)
- Lysis buffer (細胞質タンパク質用、20xを室温保存、DTT,PI,PPI,RNaseAは用事調製)
- (20mM HEPES-NaOH (pH7.5), 150mM NaCl, 2.5mM MgCl2, 0.05% Tween20, 1mM DTT, 1x PI, 1x PPI, 50ug/mL RnaseA)
- Wash buffer (20xを室温保存、DTT,PI,PPIは用事調製)
- (20mM HEPES-NaOH (pH7.5), 150mM NaCl, 2.5mM MgCl2, 0.05% Tween20, 1mM DTT, 1/20x PI, 1/20x PPI)
- 5x Elution buffer (4oC保存)
- (10mM desthio-biotin, 125mM NaCl, 25mM Tris (pHはdesthio-biotinを溶かした後、HClで7.0~7.5にあわせる))
- D-Tube Dialyzer Mini, MWCO 6-8(メルクミリポア)
- Oriole gel stain (Bio-Rad)
- SDS-PAGE gel (私の使用している既製ゲル(15分で電気泳動が完了する):e-PAGEL (MOPS buffer使用) (ATTO), Criterion TGXプレキャストゲル(Tirs-glycine buffer使用)(Bio-Rad))
- DNAゲル撮影装置(Orioleのデータ取得に使用)
おすすめ器具
- MINI ロータリー・ミキサー NRC-10D(電池稼働)(日伸理科)
- 小型ロータリーミキサーおしりぺんぺんK型NRC-20D(日伸理科)
方法
1)レジンの調製(Mag sepharose版)
1. 15 cm dish 1枚あたり5-20 μL(bed volume)のStreptavidin-Mag sepharoseを準備。レジン量はタンパク質の発現量に依存。初めは5 μLでよい。
2. 1mLのwash bufferを加え、転倒混和、マグネット (~20秒)、上澄みを除去する(洗浄)。
3. 1mLの0.1%BSA、1mM DTT入りT-bufferを加え、15分以上ローテーターでブロッキング。
4. チューブを換えながら、3回洗浄。最後の洗浄で加えたwash bufferで混和し、分注する。
5. 使用直前にマグネット (~20秒)、上澄みを除去。
6. 1日以上保存する場合、防腐剤を加える(1週間以上保存可)。
2)SBP-Streptavidinアフィニティー精製(Mag sepharose版)
1. Cellペレットに0.6~1 mL lysis bufferを加え、氷水中でPotter glass homogenizer, 20ストロークにより細胞を破砕する。
2. 15,000 x g、4oCで15分遠心
3. Option: MagでないSepharoseを使用する場合。20 μL (bed volume)のブロッキング済Sepharose 4B resin (Sigma)に上澄みを加え、4oC、1時間ローテーターでPre-clear。電池式ローテーター(おすすめ器具参照)であれば4oC冷蔵庫で実施可能。実施すると精製度が上がる。
4. 上澄みをブロッキング済Streptavidin-Mag sepharoseに加え、4oC、2時間以上ローテーターで混和する。タンパク質が失活しないことが分かっていれば、37oC、30分や室温、1時間も可。
5. マグネット (~20秒) 。
6. 上澄みを除去し、1mL wash bufferを加え、マグネットからチューブを離し、1mLシリコナイズチップを用いて新しい1.5 mLシリコナイズチューブに移す(洗浄)。3回繰り返す。タンパク質低吸着チップ、チューブを使用することで精製度が上がる。
7. 最後の洗浄後、マグネット(~20秒)、上澄みを除去。
8. Bed volumeの二倍(10~40 μL)の1 x Elution buffer (2mM desthio-biotin) with 1mM DTTを加える。必要に応じて1/20 PI, 1/20 PPIを加える。
9.氷上に30分静置(アイスオン(おすすめ器具参照)が便利)、5分毎にタッピングにより混和(溶出)。ここで、上手なヒトとそうでないヒトの違いが大きく出る。容量が200 μL以上の時は冷蔵庫で電池式ローテーター、200 μL以下の時はコールドルームで小型ロータリーミキサー(おすすめ器具参照)を用いることで実験のばらつきを解消できる(図4)。タンパク質が失活しないことが分かっていれば、室温、15分での溶出も可。
10. マグネット(~20秒)、上澄みを回収(精製タンパク質)。必要に応じて2回目の溶出。
11. 必要に応じてdesthio-biotinを透析。微量なためD-tube透析チューブを使用。
12. 精製タンパク質の精製度をSDS-PAGE後のgelをOriole gel stain (Bio-Rad)により染色することで測定。定量する場合は、BSAなどを用いて、検量線用のサンプル(1μg, 0.5 μg, 0.25 μg, 0.125 μg, 0.0625 μg, 0.03125 μg)を同時に泳動する。泳動後、DNAゲル撮影装置によりデータを取得する(図5)。CCD systemがない場合、デジタルカメラでの撮影も可。メタノールを含む染色液の廃棄に注意。
IV. その他の注意点
1. 経験的に、人工合成をおこなったcDNAは大腸菌でのコピー数が低下したり、クローニングできないことがある。コピー数の低下は、Chloramphenicol amplification of plasmids法で対応する[14]。クローニングできない場合は、薬剤濃度を下げる(Ampicillinであれば通常40-50 μg/mLを20-25 μg/mL)、大腸菌株を変更する(NEB stableがおすすめ)、ベクターの薬剤耐性を変更するなどの対応を行う。
2. サイズの大きいPlasmid、注意点1で紹介したようなPlasmidでは、容易に組み替えを起こすため、グリセロールストックを使用しない。精製毎にPlasmidを大腸菌にTransformationする。形成されたコロニーを白金時などでピックアップ後、画線し、シングルコロニー化したものを培養に用いる。
3. Kanamycin、Chloramphenicolなどのタンパク質合成阻害系薬剤を使用するTransformationの場合、プレートに塗る前にプラスミドからの薬剤耐性遺伝子発現が必須である。そのため、42oC、1分保温後、氷上に移した後に、抗生物質を入れていないLB培地(or SOC培地)を大腸菌の4倍量加え(滅菌チップ使用)、37oC 、20分培養後、アガロースプレートに塗る。
4. QIAGENなどのシリカカラムを用いた方法でのDNA精製がうまくいかない場合、通常セシウムクロライド密度勾配遠心法によるDNA大量精製を行う。最近、BACに対応したシリカカラムベースのplasmid精製キットが販売され(NucleoBond Xtra BAC)、これを用いて精製できることを確認した。従来のシリカカラムによるplasmid取得が難しい場合は試す価値がある。
5. SV40 large T antigenはGenomic instabilityを誘導する。293T、293FT細胞培養時にG418 (400 μg/mL)を加えていないと、すぐにリコンビナントタンパク質の発現量が低下する。
6. 動物細胞に発現している遺伝子のリコンビナントタンパク質を取得する際、パートナー分子の混入が起こる。どうしても除きたい場合は、低濃度Ureaによる洗浄、siRNA発現ベクターの共導入、特異抗体による免疫吸収などにより除去する[1]。
V. おわりに
紙面と時間の関係上、精製までで筆を置かしていただいた。本当は、時間の限られた技術員・共働き研究者、たくさん実験をしたい学生・ポスドクのための時短実験法をだらだらと紹介したかったのだが…。編集幹事さま、またの機会をいただけましたらありがたいです。よろしくお願いいたします!
文献
1. Arias-Palomo, E., et al., The nonsense-mediated mRNA decay SMG-1 kinase is regulated by large-scale conformational changes controlled by SMG-8. Genes Dev, 2011. 25(2): p. 153-64.
2. Melero, R., et al., Structures of SMG1-UPFs complexes: SMG1 contributes to regulate UPF2-dependent activation of UPF1 in NMD. Structure, 2014. 22(8): p. 1105-19.
3. Thomas, M., et al., Full deacylation of polyethylenimine dramatically boosts its gene delivery efficiency and specificity to mouse lung. Proc Natl Acad Sci U S A, 2005. 102(16): p. 5679-84.
4. Shimizu, Y., et al., The PURE system for protein production. Methods Mol Biol, 2014. 1118: p. 275-84.
5. Takai, K., T. Sawasaki, and Y. Endo, Practical cell-free protein synthesis system using purified wheat embryos. Nat Protoc, 2010. 5(2): p. 227-38.
6. Mikami, S., T. Kobayashi, and H. Imataka, Cell-free protein synthesis systems with extracts from cultured human cells. Methods Mol Biol, 2010. 607: p. 43-52.
7. Durocher, Y., S. Perret, and A. Kamen, High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res, 2002. 30(2): p. E9.
8. Enriquez-Harris, P., et al., A pause site for RNA polymerase II is associated with termination of transcription. EMBO J, 1991. 10(7): p. 1833-42.
9. O'Malley, R.P., et al., A mechanism for the control of protein synthesis by adenovirus VA RNAI. Cell, 1986. 44(3): p. 391-400.
10. Lopez-Perrote, A., et al., Human nonsense-mediated mRNA decay factor UPF2 interacts directly with eRF3 and the SURF complex. Nucleic Acids Res, 2016.
11. Hobel, S., et al., Polyethylenimine PEI F25-LMW allows the long-term storage of frozen complexes as fully active reagents in siRNA-mediated gene targeting and DNA delivery. Eur J Pharm Biopharm, 2008. 70(1): p. 29-41.
12. Meyuhas, O. and T. Kahan, The race to decipher the top secrets of TOP mRNAs. Biochim Biophys Acta, 2015. 1849(7): p. 801-11.
13. Nicholson, P., et al., A novel phosphorylation-independent interaction between SMG6 and UPF1 is essential for human NMD. Nucleic Acids Res, 2014. 42(14): p. 9217-35.
14. Frenkel, L. and H. Bremer, Increased amplification of plasmids pBR322 and pBR327 by low concentrations of chloramphenicol. DNA, 1986. 5(6): p. 539-44.
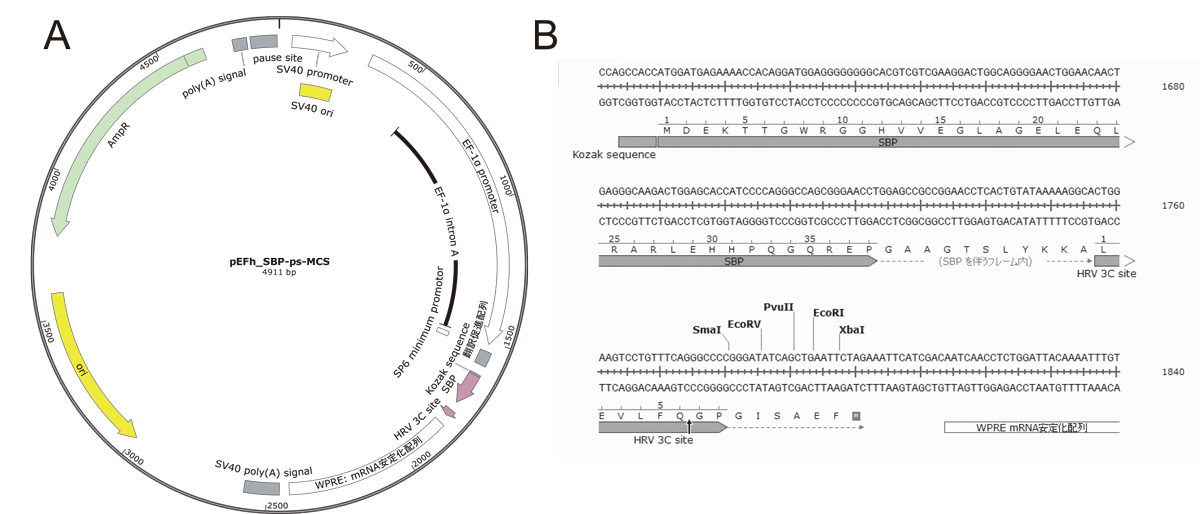 図1. ベクターの構造(A)とマルチクローニングサイト周辺配列(B)
図1. ベクターの構造(A)とマルチクローニングサイト周辺配列(B)
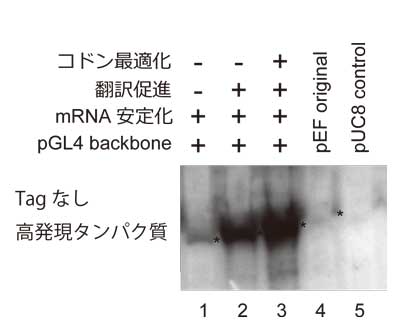
図2. mRNA転写後制御配列、コドン最適化によるリコンビナントタンパク質発現量上昇。同量のプラスミドをPEIにより導入後、全タンパク質を泳動し、高発現させたタンパク質を識別する抗体を用いてWestern blottingにより検出。Original pEFベクターの時点で内在性タンパク質の100倍以上の発現が得られている。*は発現させたタンパク質の位置。
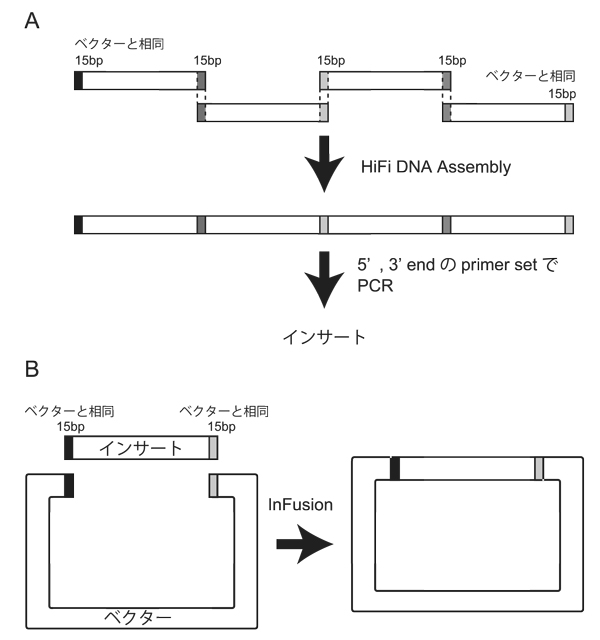
図3. HiFi Assemblyによる人工合成DNAの連結(A)とInFusionによるインサートとベクターの連結(B)
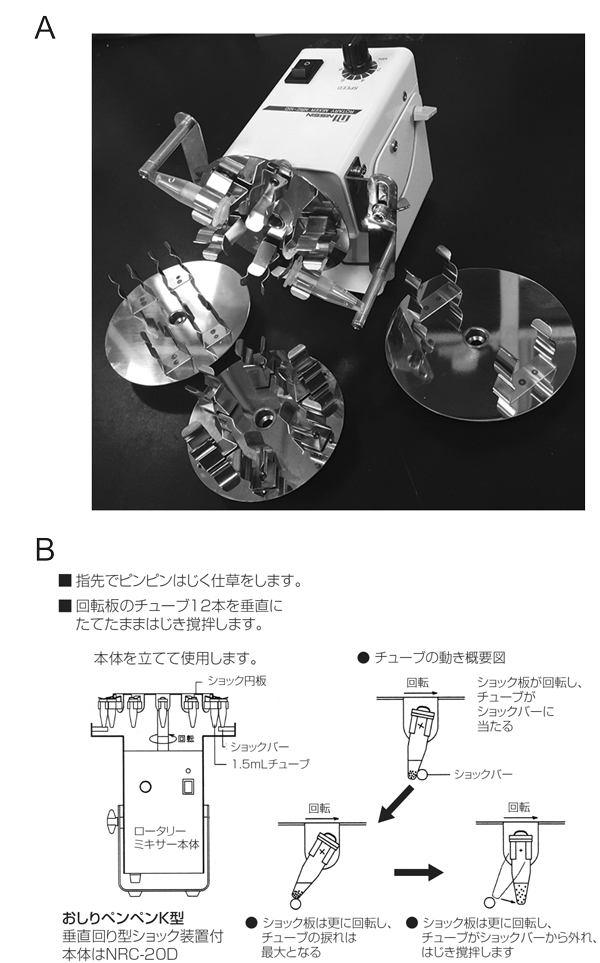
図4. (A) 電池式小型ローテーターは、最も大きなところで12 cmと小型。円盤が簡単に取り替えられ、1.5 mL ~ 50 mLチューブが使用できる。1.5 mLについては、バネ式ショック装置が付いたものがあり、レジンの攪拌を確実に行うことが出来る。(B) おしりペンペンK型は、チューブを縦にした状態で回転させ、バネ式ショック装置に当たるときにレジンが攪拌される。10 μL程度の微量な溶出も再現よく実施可能。電池式のものに比べると大きい(最大19 cm)(許可を得てカタログより転載)。
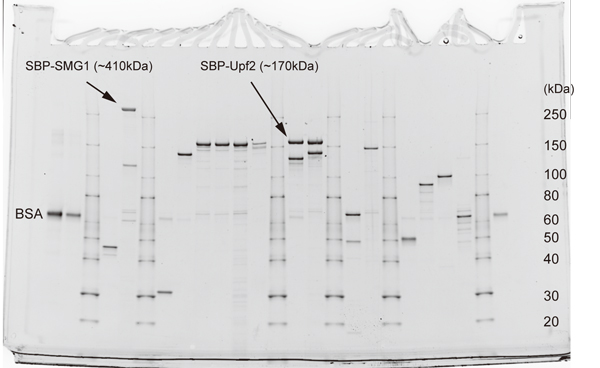
図5. 紹介した方法で精製したリコンビナントタンパク質の精製結果例。2014 Structure[2]で発表したデータのオリジナルgel。Criterion TGXでSDS-PAGEし、Orioleで染色後、BAS4000にて撮影。矢印で示したSBP-SMG1(~410kDa)は、HA-SMG8(110kDa), HA-SMG9(60kDa)と共発現させ、複合体として精製。SBP-Upf2(~170kDa)は、HA-Upf1と共発現させ、複合体として精製。



