")
")
はじめに
私は、2005年弘前にて開かれましたRNA学会年会で発表をさせて頂いたのを最後に、米国ロックフェラー大学へポスドク研究員として渡米し、実に9年ぶりに2014年の年会での発表をさせて頂きました。懇親会にて、塩見美喜子会長が「たとえばCLIPなどの最新技術についてセミナーをするなどして、RNAを扱う研究者に広くRNA学会と年会に興味をもってもらうことを考えていきたい」とお話されていたのを、乾杯用ビールを片手に隅で聞いておりました。まさか、その第一回の試みに私が寄稿させて頂くことになるとは、大変光栄に思うと同時に大役に緊張しております。
今回は、ロックフェラー大学時代に学んだHITS-CLIP法の手順について簡単にまとめさせて頂きました。2008年の発表以来、HITS-CLIP法は様々な発展と進化を遂げておりますが、最もスタンダードなものについて記載させて頂きます。
原理
CLIP法がブレークスルーとなった特徴の一つは、生きた細胞や組織をそのままUV照射する事により、蛋白質-RNA相互作用の間に共有結合させるところにある。次に、精度良く蛋白質と共有結合されたRNAを同定する手段として、適切な濃度のRNase処理により断片化し、目的の蛋白質-断片化RNA複合体を免疫沈降法による精製を行う。アイソトープを用いて断片化したRNAを標識し、電気泳動して分離した蛋白質-断片化RNAをメンブレンからアイソトープラベルを指標として切り出すことで、より目的の蛋白質に結合している特異的なRNAの精製を可能にしている。精製した断片化RNAをPCRで増幅後、次世代シークエンサーを用いてシークエンス解析する。
-準備-
■必要な試薬リスト:
Protein G (invitrogen)
RQ DNaseI (Promega M6101)
RNaseA (USB 70194Y)
CIP/CIP buffer (Fermentas Cat#EF0654)
PNK/PNK buffer (NEB M0201L)
g-ATP (パーキンエルマーBLU502A 0.25mCi 25ul)
ATP (NEB)
ニトロセルロース膜
Loading Buffer (Novex x4) non-reducing reagent (NP0007 invitrogen)
RNAsin Plus (7.5U/μl, Promega N261)
Protein K (Roche 13731196)
RNA phenol (Ambion AM9710)
3M NaOAc pH5.5 (Ambion)
glycogen(Ambion AM9510)
100 mM dNTP set (Invitrogen, 10297-018)
Superscript III (invitrogen)
Phusion®High-Fidelity PCR Master Mix with HF Buffer (NEB M0531S)
urea (Fisher, U15-3)
40% 19:1 Acrylamide:Bis-acrylamide (Fisher)
95% formamide, deionized (Sigma F-9037)
Bromophenol blue + Xylene cyanol (Sigma B-3269)
nuclease free water, 1L (AM9932)
SYBR R GOLD NUCLEI ACID GEL STAIN 500UL 10,000X CONCENTRATE IN DMSO (InvitrogenS11494)
Nanosep MF Centrifugal Devices (0.45 µm, 100PK) (VWR 29300-644)
RT-PCR Grade Water (10 x 1.75 ml) (Ambion AM9935)
■使用するバッファー:
・1xHBSS
・Wash buffer
1xPBS
0.1% SDS
0.5% deoxycholate
0.5% NP-40
・High-salt wash buffer
5xPBS
0.1% SDC
0.5% deoxycholate
0.5% NP-40
・1xPK buffer
100 mM Tris-HCl pH7.5
50 mM NaCl
10 mM EDTA
・1xPK buffer/7M urea
100 mM Tris-HCl pH7.5
50 mM NaCl
10 mM EDTA
7M urea
・1xPNK buffer
50 mM Tris-HClpH7.4
10 mM MgCl2
0.5% NP-40
・1xPNK + EGTA buffer
50 mM Tris-HCl pH7.4
20 mM EGTA
0.5% NP-40
・2x Loading buffer
95% formamide, deionized (Sigma F-9037)
5% 100mM EDTA, pH 8
Bromophenol blue + Xylene cyanol (Sigma B-3269)
■使用する機械:
・ Thermomixer R (エッペンドルフ社)
・ UV Stratalinker (Stratagene現在発売中止、その他可)
・卓上超遠心器(Beckmann社)
■使用するRNA リンカー(グライナージャパンなど):
RL5: 5’-OH-GUUCAGAGUUCUACAGUCCGACGAUCNNNNG-3'-OH
RL3: 5’P-UCGUAUGCCGUCUUCUGCUUG-3’-amino acid
(参考)
例:バーコード付き5’リンカー
RL5-AGCT 5’-OHGUUCAGAGUUCUACAGUCCGACGAUCAGCUUNNNNG-3'-OH
RL5-ATGC 5’-OH GUUCAGAGUUCUACAGUCCGACGAUCAUGCUNNNNG-3'-OH
RL5-CAGT 5’-OH GUUCAGAGUUCUACAGUCCGACGAUCCAGUUNNNNG-3'-OH
RL5-GTCA 5’-OH GUUCAGAGUUCUACAGUCCGACGAUCGUCAUNNNNG-3'-OH
RL5-TGAC 5’-OH GUUCAGAGUUCUACAGUCCGACGAUCUGACUNNNNG-3'-OH
■使用するDNA プライマー(北海道システムサイエンスなど):
DSFP5-PE:
AATGATACGGCGACCACCGAGATCTACACCAGGTTCAGAGTTCTACAGTCCGACG
Read1F:
CAGGTTCAGAGTTCTACAGTCCGACG
Reverse:
SP3: CAAGCAGAAGACGGCATA
プロトコール
❶ 細胞、あるいは組織のUVクロスリンク(Day1)
1)組織の場合
①10 mlピペットを用いて10x volumeのHBSSバッファーで軽く分散する。(約3回のピペッティング)
②200 mlピペットtipを先に付けた10 mlピペットを用いてさらに組織を分散する。(約3回のピペッティング)
③氷の入ったトレイ中に置いた10 cm ディッシュへ組織/細胞を移す。
④ディッシュのフタを開けた状態でStratalinkerにより3回UV照射する(100-400 mJ/cm2)。この時、UV照射毎に毎回ディッシュを揺する。
⑤細胞を2500 rpm、5分で遠心後、上清を除きエッペンチューブに1mlのHBSSで細胞を回収する。
⑥5000 rpm、2分で遠心後上清を除いてペレットの状態で、-80℃に保存する。
2)培養細胞の場合
①10cm ディッシュで培養した接着細胞を6 mlのice cold PBSでリンス後、2回UV照射(100-400 mJ/cm2)する。以降の操作は1)の⑤⑥に同じ。
* コメント
目的のRNA結合蛋白質により、一次構造を認識するものや高次構造を認識するようなものがありUVクロスリンクの強度はそれぞれ検討の必要がある。最大シグナル強度の70%以上のシグナルが得られる最も少ないUV量を至適として用いる。単層培養細胞へのUV照射は目安として組織の半分程度でよい。
❷ RNase消化と免疫沈降(Day2)
1)ビーズ準備
①1サンプルに対して50 mlのprotein GあるいはA DynabeadsをPBS 0.02% Tween20 で3回洗浄する。
②目的の蛋白質の抗体を計400 ml(PBS 0.02% Tween20)になるようにビーズに加える。(室温で45分間、転倒撹拌)
③PBS 0.02% Tween20で3回洗浄する。(要検討:ここでのBufferは用いる抗体に依存する。Dynabeadsのユーザーガイド参照)
2)RNaseによる部分消化
①UVクロスリンクしたサンプルペレットをWash bufferで溶解する。(10分間氷上)
(特に溶解に使うBufferは用事調整が望ましい)
②RQ1 DNAse (Promega, M6101)を15 mlをそれぞれのサンプルに加える。
(37℃で5分間震とう撹拌1000 rpm, Thermomixer (Eppendorf))
③RNAse A (USB 70194Y) 1:100-20,000に希釈したRNase A (USB 70194Y)を5 ml加える。(37℃で5分間震とう撹拌1000 rpm)
④超遠心(4℃,30,000 rpm, 20分間)で上清を得る。 (polycarbonate tubes in TLA 120.2 rotor)
3)免疫沈降
①上清とBeadsを混ぜる。(4℃, 1-4時間、転倒撹拌)
②Wash buffer、High-salt wash buffer、1xPNK bufferでそれぞれ2回づつ洗浄。
(Washは、毎回3-5分rotationする)
4)CIP処理
①CIP反応液を調製する
CIP反応液:
2 µl 10x dephosphorylation buffer (Roche, 712023)
0.75 µl alkaline phosphatase (Roche, 712023)
0.2 µl 10% Tween-20
17.05 µl water
(20 µl total)
②37℃で20分間、撹拌震とう(1200 rpm, 2 分ごと 15 秒間)後、PNK buffer1回、PNK+ EGTA buffer、PNK buffer2回の順でビーズを洗う(Washは、毎回3-5分rotationする)。
❸ 3’リンカーライゲーション
①それぞれのチューブに20 mlのlinker mixを加える。
Linker mix:
4 µl RL3 linker 20 pmol/µl
16 µl water
(20 µl total)
②さらに、それぞれのチューブに20 mlのligase mixを加える。
Ligase mix:
4 µl 10X T4 RNA ligase buffer (Fermentas)
4 µl BSA (0.2 µg/µl)
4 µl ATP (10 mM)
1 µl T4 RNA ligase (Fermentas)
7 µl water
(20 µl total)
③16℃で一晩、撹拌震とう(1200 rpm、1.5 分ごと 15 秒間)する。
* コメント
リンカーRNAは、変性gelで泳動後、切り出し精製したものを使う。-80℃で保存、凍結融解は繰り返さないこと(核酸切り出し手順は、最後のページを参照)。今回はリンカーRNAにamino acidを標識しているが、リンカー同士のライゲーションを避けるたに、他の修飾でも構わない。
❹ PNK処理とSDS-PAGE/転写(Day3)
①PNKbuffer、High-salt Wash buffer 2回、PNK bufferの順でビーズを洗う。
(Washは、毎回3-5分rotationする)
②40 mlのPNK mixを加え、37℃で20分間、撹拌震とうする(1200 rpm 2 分ごと 15 秒間)。
PNK mix:
4 µl 10X PNK Buffer (NEB)
2 µl 32P-γ-ATP
2 µl T4 PNK enzyme (NEB, M0201L)
32 µl water
(40 µl total)
③1mM ATP 5 mlを加え、さらに5分間震とうする。
④PNK bufferで1回、High-salt Wash buffer 1回、PNK bufferで2回の順でビーズを洗う。(Washは、毎回3-5分rotaitionする)
⑤15 µlの1X PNKと15 µl 4x Novex loading bufferを加え、70℃で10分間震とう撹拌後、マグネットスタンドを使って上清を回収する。
⑥Novex NuPAGE 10% Bis-Trisゲルにサンプルをアプライして、4℃で175Vの定電圧で電気泳動する。(約1時間)
⑦電気泳動後、ニトロセルロース膜にゲルを30Vの定電圧で転写する。
⑧転写後、ニトロセルロース膜をPBSでリンス後、ラップに包み、オートラジオグラムで露光する。
⑨フィルム上のシグナルを指標に目的のタンパク質−RNA断片を外科用メスを用いて切り出す。
* コメント
RNaseの条件検討を本実験の前に行う。目的の蛋白質よりスメアに10〜20kDaくらいシフトする条件が至適RNase濃度となる。少なくとも、High RNaseとLow RNase条件のサンプルを泳動し、比較する。またHigh RNase処理した条件で予想される蛋白質の分子量サイズの上にシグナルがないことも確認する(コンタミを避ける)。コントロールとして、UV照射なし、コントロールIgGなどのレーンも用意する。それでもバックが気になる場合は、細胞抽出液をコントロールIgG-Dynabeadsでプレインキュベーションした上清を目的の抗体での免疫沈降に使用する、あるいはDynabeads自体をBSAなどでコートする等の工夫を行ってみるとよい。また、通常のSDS-PAGEゲルでは、pHがアルカリ性になることでRNAが加水分解されてしまうため、市販のinvitrogenプレキャストゲルを使うのが好ましい。
❺ RNAの単離(Day4)
①使用前に4mg/ml proteinase K (Roche, 1373196) を含む1X PK Bufferを予め、37℃ 20分間、加温する。(内在性のRNase活性を抑えるため)。
②それぞれ砕いたニトロセルロース膜の入ったチューブに200 µlずつ加える(37℃, 20分, 1000 rpm)。
③200 µl の1x PK/7M urea溶液を加える。(37℃, 20分, 1000 rpm) (c.f 用事調製: 1.05 g Urea/2.5ml)
④次に400 µl RNA phenol (Ambion, 9710)と130 µl of CHCl3 (chloroform 49:1 with isoamyl alcohol)を加える。(37℃, 20分, 1000 rpm)
⑤フルスピードでチューブを遠心後(室温 5分)、上清に50 µl 3M NaOAc pH 5.5と0.75 µl glycogen (Ambion, 9510),1ml of 1:1 EtOH:isopropanol を加える。(一晩, -20℃)
*コメント
できるかぎり、カットするメンブレンの大きさは、3kDaサイズにカットし、さらにそれを細かく砕く。
❻ 5’リンカーライゲーション(Day5)
①RNAをフルスピードで遠心(4℃, 10分)
②75% EtOH 150 µl を加えて洗浄/ボルテックス
③フルスピードでチューブを遠心後(4℃, 10分)
④75% EtOH 1mlで再度洗浄する
⑤フルスピードでチューブを遠心後(4℃, 10分)
⑥ペレットを5.9 µl H2Oで溶解する。
⑦計10 mlになるようにRNA ligation mixを加える(16℃, 1-5時間 or 一晩)
RNA ligation mix:
1 µl 10X T4 RNA ligase buffer (Fermentas)
1 µl BSA (0.2 µg/µl)
1 µl ATP (10 mM)
0.1 µl T4 RNA ligase (3U, Fermentas)
1 µl RL5 RNA linker 20 pmol/µl
⑧ライゲーション反応液に79 µl water, 11 µl 10X DNase Iバッファー、5 µl RNAsin, 5 µl RQ1 DNAseを加える 。(37℃, 20分)
⑨300 µl water, 300 µl RNA phenol、100 µl CHCl3 を加えフェノール処理を行う。
⑩上清を回収後、50 µl 3M NaOAc pH 5.5, 0.5 µl glycogen (Ambion, 9510), 1 ml 1:1 EtOH:isopropanol を加える。(-20℃ 一晩 or 1hr)
* コメント
塩の析出や過度のペレットの乾燥はライゲーション効率を大きく妨げる可能性があるため再度フェノクロ処理をすることを薦める。(以降の実験で、リンカー-リンカーライゲーション産物が得られた場合、ここのステップに入る前に、RT反応に進む前に変性ゲル上で目的のサイズのRNAライブラリーをゲル精製するステップを挟む事をお薦めする。RIラベルを指標にサイズの確認も可能)
❼ RT-PCR(Day6)
1)RT反応
①RNAをフルスピードで遠心(10分)、75% EtOHリンス後、ペレットを乾かし、8 µlの水に溶かす。次に、2 µlのSP3 (5 pmol/µl)と3 µlの3 mM dNTPsを加えて65℃、5分熱処理する。
②RT反応液を調製する
RT反応液:
1 µl 0.1 M DTT
4 µl 5X SuperScript RT Buffer
1µl RNAsin
1 µl SuperScript III (Invitrogen 18080-044)
(total 20 µl)
③RT反応、50℃45分間、55℃15分間、90℃5分間の後、4℃インキュベートする。
2)1st PCR反応
①RT反応物を鋳型とするPCR反応液を調製する
PCR反応液:
7.5 µl 2xPhusionHF (NEB: M0531S)
0.25 µl Read1F primer, 20 pmol/µl
0.25 µl SP3 primer, 20 pmol/µl
2 µl Water
5 µl RT産物
(total 15 µl)
②PCR 98℃ 30秒,18-24サイクル(98℃10秒/ 60℃10秒 / 72℃ 10秒), 72℃1分
*コメント
サイクル数をふっておくこと。(目安: 18-24サイクル等)
例えば、上記PCR反応液中に50x希釈SYBR Green I(Invitrogen, S7563)を0.5 µl加え、リアルタイムPCRマシーンで増幅カーブを確かめながらサイクル数を決定する事も可。これによりテンプレートcDNAの節約が可能。また-RTコントロールも取る事、半量あるいは1/4量を-RTに回した場合、サイクル数をその分-RTコントロールは多く取る。
❽ イルミナシークエンス(Day7)
①10% 変性ポリアクリルアミドゲルでPCR産物を電気泳動し、TBEで10,000倍希釈したSYBR Gold (Molecular Probes)でゲルを10分間染色し、DNAをチェックする(LEDライトがお薦め)。
②100-130 ntサイズのDNAを切り出し,ゲルを精製する。(PCR産物がラダーになったり、シングルバンドのようなものが見える場合は、リンカーリンカーライゲーションのような産物が疑われる)
*コメント
目的の蛋白質の〜20kDa上のバンドを3kDa程の大きさで切り出した場合、PCRのバンドは120〜150ntに見える。
③2nd PCR反応
PCR反応液:
7.5 µl 2xPhusionHF (NEB: M0531S)
0.25 µl DSFP5-PE primer, 20 pmol/µl
0.25 µl SP3 primer, 20 pmol/µl
2 µl Water
5 µl 1stPCR産物
(15 µl total)
④PCR。98℃ 30秒, 4-8サイクル(98℃10秒/ 60℃10秒 / 72℃ 10秒), 72℃1分
⑤PCR産物は、AMPure XP Beadsを用いて精製を行う。
⑥DNA濃度をBioanalyzer(アジレント社)Qubit(ライフテクノロジーズ)、qPCRアッセイで調べた後、イルミナシークエンスする。
■ 核酸のゲル精製
・核酸溶出buffer
1M NaOAc (pH 5.5)
1mM EDTA
・2X Formamide Loading buffer
95% Formamide, deionized (Sigma, F-9037)
5% 100mM EDTA, pH 8
Bromophenol blue + Xylene cyanol (Sigma B-3269)
・変性ゲル(total 20ml)
8.4 g Urea
5 ml 40% 19:1 Acrylamide:Bis-acrylamide (ナカライ)
4 ml 5x TBE
100 ul 10%APS
15 ul TEMED
プロトコール
①切り出した核酸に350mlの核酸溶出bufferを加え; 1ml シリンジのプランガーを用いて、ゲルを粉砕する
②37°C 30 分間、震盪する1200 rpm (サーモミキサー)
③ゲル溶液を、Nanosep MF columnに加える(1cm glass pre-filterを敷く, ワットマン社1823-010)
④フルスピードで1分間遠心後、フロースルーを回収
⑤1ml 1:1 EtOH:Isopropanol溶液を加える(2 時間/ ON at -80°C / -20°C)
⑥スピンダウン(14,000rpm for 15 分, 4°C)
⑦75% エタノールで2回洗浄
⑧適切な濃度になるように水で回収する。
おわりに
これまで私は、一貫して神経系におけるRNA結合蛋白質の機能解析を行ってまいりました。私の大学院時代2001-2005年というのは、ゲノムプロジェクトの終了、RNA新大陸の発見があり、新しいポストゲノム科学に大きな期待が寄せられていました。一個人、研究者としては、これらの大発見は、無限のように思えた生命の設計図が、実はゲノムDNAを有限なものとして捉えることが可能であるという興奮、と同時に想像以上にRNAによる複雑系を目の前にして気が遠くなる思いもしました。その中で、私の博士研究員時代(2006-2011年)の師であるRobert B. Darnell博士(ロックフェラー大学教授、現在、NYゲノムセンターCEO兼任)は、生きた動物、組織や脳そのものを、生化学的に扱う事で、正確に、限りあるゲノム情報から産生されるRNAの複雑系を分子レベルで(ひいては1塩基解像度、1オングストロームの解像度で!)トランスクリプトームワイドにひも解くという新しいストラテジーで研究の発展に成功させました。HITS-CLIP法は、皆様ご存知のように強力な網羅的、蛋白質—RNA相互作用の同定法として広く認識された事は言う迄もありません。それでもそれ以上に、研究者たちの興味、関心を引く理由を考えてみると、トランスクリプトームワイドレベルでの定量的解析にあると思います。非コードRNA研究を含めたRNAの分野では、新しい概念が提唱され、その生物学的意義にも注目が集まっております。では、その現象をin vivoで化学量論的に実証できるのか、と考えた時にやはりHITS-CLIPのようなin vivo生化学的な証明が必要となります。例えば、Loeb GBらのMol.cellの論文では、特定のmiRNA(miR-155)の欠損マウスを用いたd-CLIP解析(ディファレンシャルCLIP)で正確なmiRNA標的mRNAが同定されています。また、この論文の考察をじっくり読むとceRNA(competing endogenous RNA)概念の定量的理解について述べられていたりすると、HTS-CLIPが改めてとても味わい深いものに感じる次第であります。
筆者プロフィール
矢野真人:2014年12月より新潟大学大学院医歯学総合研究科神経生物・解剖学分野(解剖学第二)准教授.
1999年東京工業大学生命理工学部卒業、2005年大阪大学医学系研究科博士課程卒業. 医学博士. 慶應義塾大学医学部生理学教室(岡野栄之教授研究室)日本学振振興会特別研究員PD、2006年ロックフェラー大学研究員、日本学振振興会海外特別研究員を経て、2011年慶應義塾大学医学部生理学教室に帰室. HITS-CLIP法にを用いて、神経系の発達・疾患・進化の謎に定量的、多面的に、そしていかに生物学的に迫れるかチャレンジしたいと考えております。何か質問等がございましたら、私まで問い合わせて頂けましたら幸いです。
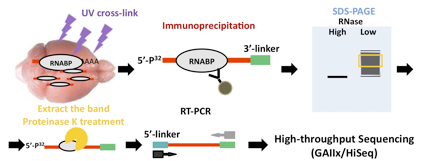 図1:HITS-CLIP法の手順
図1:HITS-CLIP法の手順
この図は、慶應義塾大学岡野研究室へ帰室後、最初に一緒に仕事をした元大学院生の陶山智史君(現、塩野義製薬)が作製。この場を借りて謝辞を述べさせて頂きます。



